


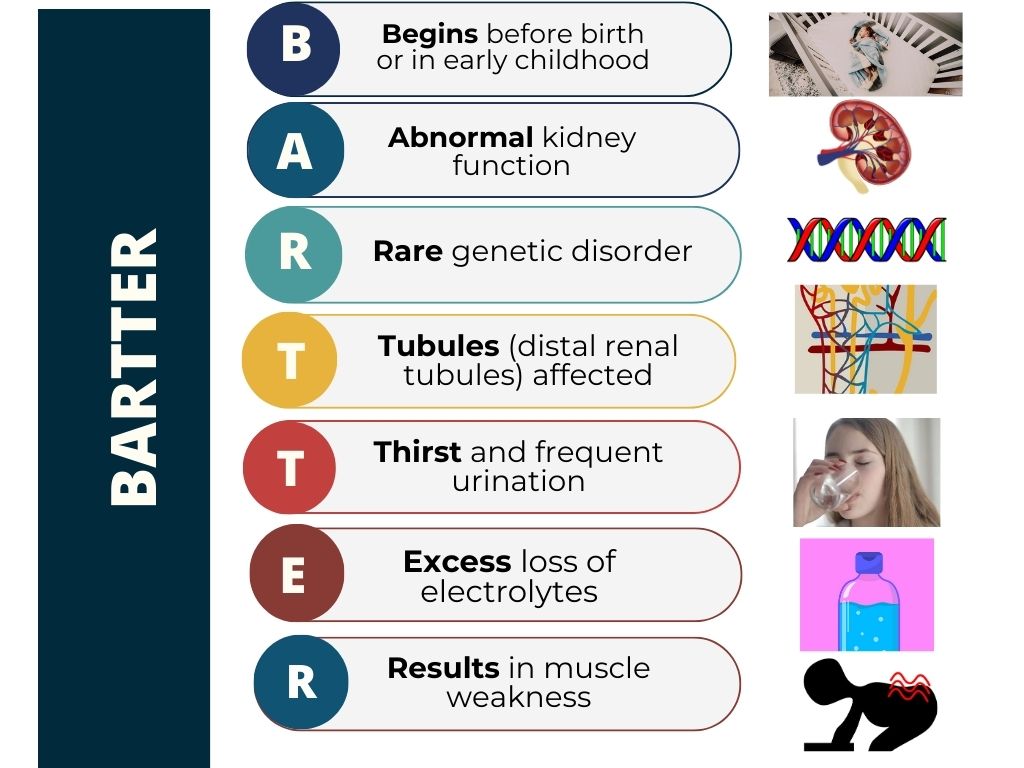
Bartter syndrome can be broken down into the following features:
Created by: Deepika Rana
Bartter syndrome is a series of rare genetic illnesses that cause deficits in kidney function. These defects affect the kidney's capacity to reabsorb salt, resulting in unbalanced body fluid and electrolyte levels.
Bartter syndrome is challenging to treat, and there is no known cure. Leaving Bartter syndrome untreated creates a high risk of the condition worsening, even potentially causing death, with chronic renal (kidney) disease playing a significant part in these outcomes. This article will assist you in giving the appropriate information and treatment advice.
Overview
Bartter syndrome is a condition that affects the ability of the kidneys to reabsorb salt and other electrolytes. This leads to excessive loss of electrolytes in the urine. Low blood pressure, difficulty reabsorbing salts, and depletion of extracellular fluid volume are the main features of Bartter syndrome. The syndrome is genetic (known as autosomal recessive), meaning it is inherited at conception. Both parents must possess and pass the gene onto their child for clinical signs to manifest.
Electrolytes affected are mineral salts like potassium, calcium, magnesium, sodium, and chloride. Numerous electrolyte abnormalities, such as low potassium (hypokalemia), low chloride levels (hypochloremia) and, in rare instances, low magnesium (hypomagnesemia), are defining features of the condition.
Other problems associated with Bartter syndrome include:
- Elevated hormone levels, including prostaglandin E2 and secondary hyperaldosteronism
- Excessive renin enzyme levels
- Metabolic alkalosis (imbalanced body fluid pH)
- Failure to thrive in infancy
The kidney consists of several structures - the most important of which, in Bartter syndrome, is called the loop of Henle, which transports water, urine, and waste products within the kidney. The thick ascending limb of the loop of Henle (TALH) is responsible for reabsorbing sodium and chloride ions (salt) to help the body maintain a functioning water-electrolyte balance. In Bartter syndrome, this function is disrupted and contributes to the loss of several other electrolytes and minerals in urine, including:
- Potassium
- Hydrogen ions
- Calcium
- Magnesium
Patients with Bartter syndrome also frequently have nephrocalcinosis - where too much calcium is deposited in the kidneys, alongside excessive loss of calcium in the urine.1
Dr Frederic Bartter first described the disease in the 1960s, stating that the age at which visible signs first appear might range from before birth to adulthood and that a change in one of several distinct genes may cause Bartter syndrome.
Causes and subtypes of bartter syndrome
Mutations in at least five genes are associated with the development of Bartter syndrome. The following table shows various subtypes with the defective gene:
| Genetic Type | Defective Gene | Clinical Type |
| Type I | The gene NKCC2 (locus SLC12A1 on chromosomal bands 15q15-21) encodes a sodium/potassium chloride cotransporter. | Neonatal (symptoms typically appear during this period) |
| Type II | ROMK gene (locus KCNJ1 on chromosome bands 11q24-25) | Neonatal |
| Type III | Chloride-channel gene (CLCNKB, on band 1p36) The inability of chloride to exit the cell inhibits the sodium/potassium chloride cotransporter. Volume constriction, hypokalemia, and high angiotensin levels all lead to a rise in intrarenal prostaglandin E2 (PGE2) synthesis, which feeds a vicious loop by preventing sodium chloride reabsorption in the TALH and enhancing the renin-aldosterone axis. | Classic (symptoms are commonly associated with Bartter syndrome) |
| Type IV | BSND encodes barttin. The deficiency hinders the barttin-dependent insertion of the chloride channel subunits ClC-Ka and ClC-Kb in the plasma membrane leading to mutations in the barttin gene, causing sensorineural deafness. | Neonatal deafness (symptoms typically appear during this period and include hearing loss) |
| Type IVb | CLCNKB and CLCNKA | Neonatal deafness |
| Type V | Digenic disorder, deletion in the CLCNKB gene and a point mutation in the CLCNKA gene. A gain-of-function mutation in the calcium-sensing receptor (CaSR) is the cause of type V Bartter syndrome. This receptor's activation decreases the rate of potassium efflux from the ROMK channel, which reduces the activity of the Na-K-2Cl cotransporter. The lack of luminal positive charge increases calcium and magnesium levels in urine.3 | Classic |
Signs and symptoms
According to the various stages of a child's growth, there are distinct signs and symptoms of Bartter syndrome. These include:
Before birth:
- Polyhydramnios (accumulation of amniotic fluid in the uterus during pregnancy)
Infants:
- Premature birth
- Dehydration
- Polyuria (frequent and excessive urination)
- Rapid weight loss
- Sensorineural deafness (deafness often caused by inner ear damage)
Older infants and young children:
- Constipation
- Polyuria and polydipsia (excessive thirst) leading to hypovolemia (loss of body fluids)
- Cramping
- Vomiting
- Stunted growth
- Failure to thrive
- Salt craving
- Muscle weakness
- Low blood pressure
- Unexplained fever
- Delayed growth
In older children and adolescents:
- Thirst
- Fatigue
- Nocturia (frequently needing to urinate at night)
- Poor growth with pubertal delay
- Constipation
- Thin face with a prominent forehead
- Large eyes and strabismus (crossed eyes)
- Protruding ears
- Sensorineural deafness
- Drooping mouth
- High blood pressure
In rare cases, children can be born without symptoms, making diagnosis difficult. These individuals may get diagnosed by genetic screening when there is a known family history of the illness or by blood or urine testing. They often present with blood samples that are highly alkaline and low in potassium. Furthermore, calcium salt deposits may be found in the kidneys (nephrocalcinosis).1,2,3
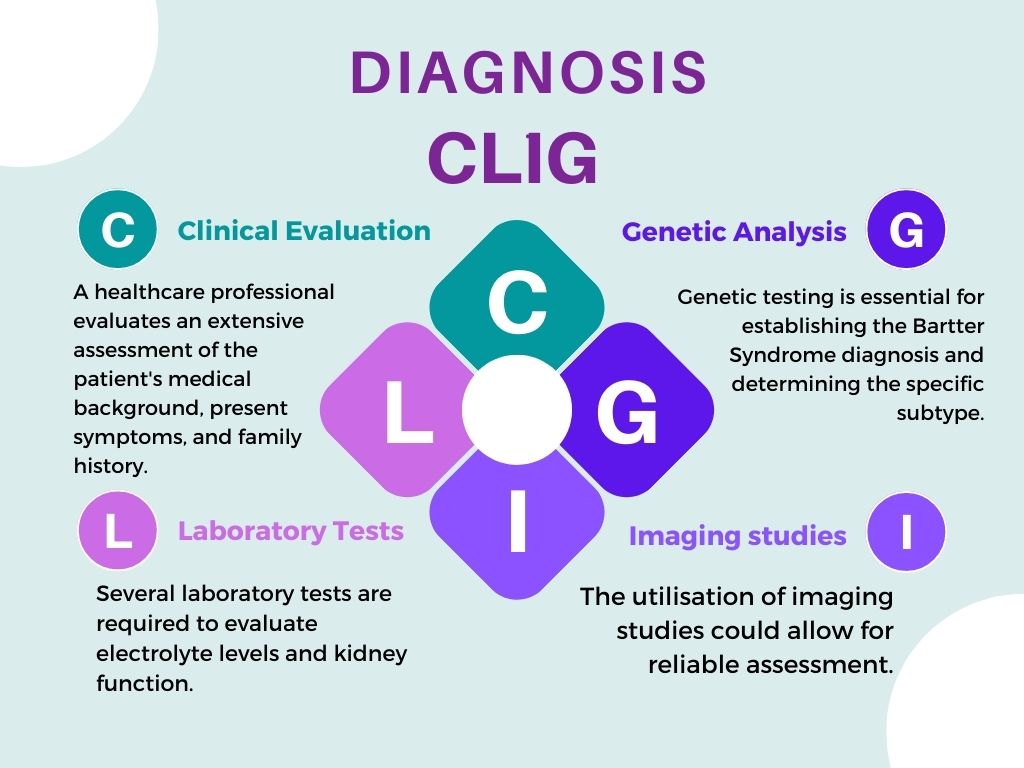
Diagnosis
A thorough evaluation is required to correctly identify Bartter syndrome. This is due to the rarity of the disorder and the diversity of how it presents from person to person.
Created by: Deepika Rana
Laboratory tests
- Urine analysis - doctors will look for elevated levels of sodium, potassium, chloride, magnesium, calcium and PGE2 hormone excretion in urine, along with elevated renin (enzyme) and aldosterone (hormone) levels
- Complete blood count - patients with Barrter syndrome may have excessive circulating red blood cells (polycythemia)
- Blood pressure - patients may have low blood pressure due to hypovolemia (an excessive fluid loss that causes weakness and dizziness)
- Serum phosphate and parathyroid hormone test - low serum phosphate levels can be prevalent among patients, particularly those with type III Bartter syndrome, due to the kidneys getting rid of too much phosphate in the urine
- Biochemical analysis of amniotic fluid - crucially performed for a prenatal diagnosis of Bartter syndrome (Gitelman syndrome and Bartter syndrome both have a higher-than-normal chloride concentration and there have also been reports of low total protein levels and elevated levels of aldosterone in amniotic fluid)
- Renal function test - the glomerular filtration rate (GFR), which controls the fluid-electrolyte balance, remains steady in the early stages of the illness, but prolonged hypokalemia can cause a decline in GFR1
Imaging studies
- Renal ultrasound - monitoring individuals with Bartter syndrome and nephrocalcinosis during anti-inflammatory therapy is often done via renal ultrasound (usually used for detecting kidney stones or calcium salt deposits)
- Computed tomography (CT scan) - although a CT scan offers a more accurate assessment of renal calcium deposits than renal ultrasound, it is associated with increased radiation. It should only be used in clinical settings when it provides a clear therapeutic advantage, such as locating stones in obstructive uropathy, which can occasionally occur in Bartter syndrome.2,4
Genetic analysis
- In Bartter syndrome, there may be multiple genetic changes. Using genetic testing, doctors will look for missense/non-sense mutations, splicing substitutions, minor deletions, and extensive deletions, particularly in the SLC12A1, CLCNKB, and KCNJ1 genes
- Ultra-sequencing or Next Generation Sequencing (NGS), typically scans for harmful alterations of known disease-linked genes3,5
Treatment and management
The primary objectives of Bartter syndrome treatment are to reduce symptoms, address electrolyte imbalances, and enhance overall quality of life. Since this is a chronic, long-term illness, continual medical attention and supervision are crucial.
Electrolyte replacement
Potassium chloride supplementation can help treat hypokalemia (excessive potassium loss), but symptoms may persist despite being less severe. Excess potassium loss can also be stopped by treatment with ‘potassium-sparing diuretics’ that increase urination without the loss of potassium.
Angiotensin-converting enzyme (ACE) inhibitors are another type of medication employed to treat high levels of protein in urine (proteinuria) or raise low potassium ion levels. Due to the prevalence of excessive urination and dehydration in this high-risk patient population, caution is encouraged due to the potential risk of injuring the kidneys.
NSAIDs (nonsteroidal anti-inflammatory drugs)
NSAIDs help to decrease urine output, therefore resulting in less electrolyte loss and helping to increase, for example, low blood potassium.
Kidney transplants
According to reports, when patients with end-stage renal disease (ESRD) due to Bartter syndrome receive a kidney transplant from a matched relative, their hormone and, therefore, electrolyte levels exhibit an improved balance after the procedure.
Monitoring growth and development in children
Paediatricians will monitor the growth and development of children with Bartter syndrome to ensure appropriate interventions are utilised. For example, treatment for low height may involve the administration of growth hormone (GH).
Genetic counselling
Genetic counselling is essential for individuals with Bartter syndrome and their families, as they should be aware of information on the inheritance pattern of the disease, be able to receive a risk assessment for upcoming pregnancies and be advised on family planning alternatives.1,6
Complications and long-term outlook
The primary complications of Bartter syndrome are:
- Sensorineural deafness
- Calcium salt formations in the kidney (nephrocalcinosis)
- Kidney failure
- Decreased/slowed growth resulting in short stature
If Bartter syndrome is left untreated, significant worsening of the disease and increased chance of death can occur. Disease prognosis improves substantially with treatment. However, long-term monitoring and management are essential due to the slow advancement of chronic renal failure.
Additional complications can include:
- Sudden death and cardiac arrhythmia caused by imbalances in electrolytes
- Delayed development and failure to thrive in untreated patients
- Decrease in bone mineral density observed in patients with either the neonatal or classic form6
Despite ongoing research, how the condition presents at different stages and in different people is still not completely known. As a result, there are still a lot of unanswered questions regarding how this disease progresses and any other potential side effects. Therefore, it is essential to use specific diagnostic techniques to make precise clinical assessments in order to avoid false conclusions and improve patient outcomes.5
FAQs
How common is bartter syndrome?
Bartter syndrome occurs in approximately 1 in 1,000,000 people worldwide, making it a rare disorder.1
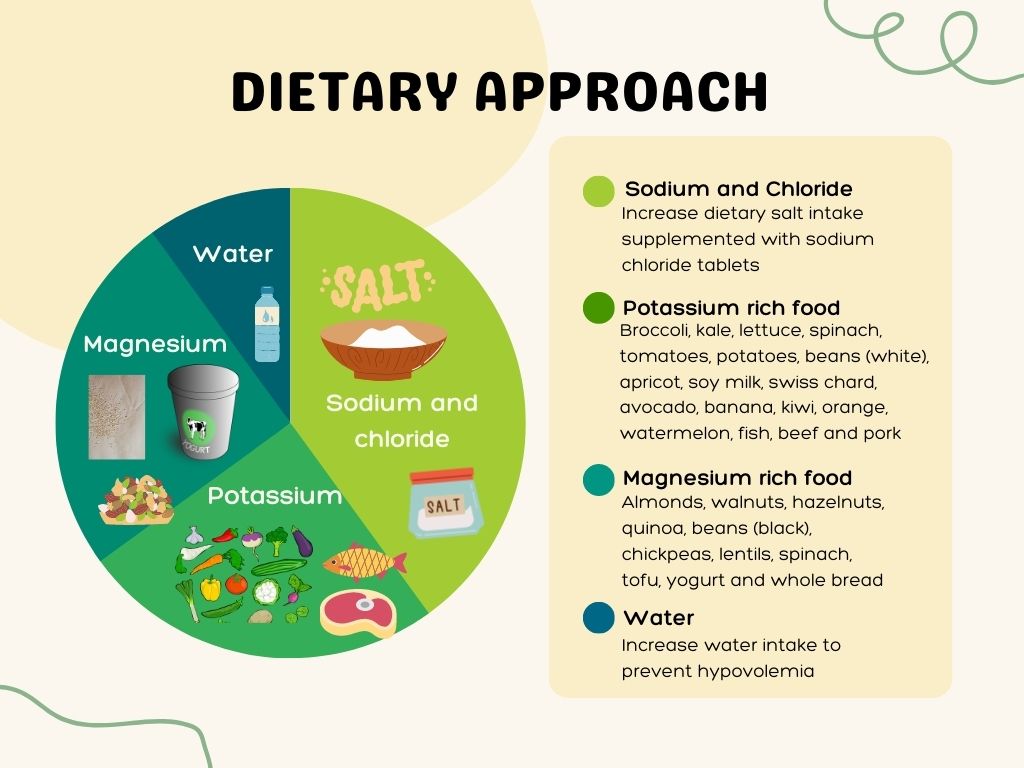
Are there any dietary changes recommended for people with bartter syndrome?
An electrolytes-rich diet is advised to compensate for the losses caused by Bartter syndrome.8
Created by: Deepika Rana
Does bartter syndrome affect pregnancy?
Bartter syndrome causes a significant decrease in creatinine clearance, exposing pregnant patients at risk for pregnancy-induced hypertension, concurrent foetal growth limitation, and oligohydramnios. It is necessary to supplement with more potassium and sodium due to the increased plasma volume typically associated with pregnancy and the kidney's inability to maintain electrolyte balance due to this condition.7
Can Bartter syndrome be mistaken for other conditions?
The symptoms of Bartter syndrome can sometimes be mistaken for other kidney-related or metabolic disorders, such as:
- Gitelman syndrome - individuals with the condition experience fatigue, muscle weakness, pains, cramps, and spasms
- EAST syndrome - the underlying abnormality that results in salt and electrolyte imbalances in EAST syndrome is similar to Gitelman and Bartter syndrome
- Cystic fibrosis - Young children with certain risk factors (such as severe respiratory or pancreatic disease, gastrointestinal losses, or warm temperatures) may experience symptoms resembling Bartter syndrome
- Pseudo-Bartter syndrome is a term used to describe several illnesses that have similar signs and symptoms to Bartter syndrome but do not involve inherited renal tubular failure
- Autosomal dominant hypocalcemia type 1 (familial hypocalcemia)
Summary
In summary, this article provides an exploration of Bartter syndrome, a rare genetic kidney disorder. The genetic basis, clinical manifestations, and diagnostic approaches were examined, emphasizing the significance of genetic testing in identifying specific subtypes. Early diagnosis and interventions, such as potassium supplementation and potassium-sparing diuretics, are crucial for symptom management. Although treatment improves the prognosis, the long-term outlook is guarded due to the risk of chronic renal failure. Ongoing genetic medicine research offers hope for enhanced understanding and improved therapeutic approaches for this uncommon yet impactful condition. Individuals and families affected by Bartter syndrome can work closely with medical specialists to properly manage the disorder and enhance their overall quality of life by understanding its causes, symptoms, and potential therapies.
References
- Bokhari SRA, Zulfiqar H, Mansur A. Bartter syndrome. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 Aug 7]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK442019/
- Konrad M, Nijenhuis T, Ariceta G, Thomas AB, Calo LA, Capasso G, et al. Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders.[ Internet] Kidney International (2021) 99, 324–335 [cited 2023 Aug 9]; Available from: https://www.erknet.org/fileadmin/files/user_upload/Bartter_Syndrome_consensus_paper__Kidney_Int_.pdf
- Florea L, Caba L, Gorduza EV. Genetic heterogeneity in Bartter syndrome: clinical and practical importance. Frontiers in Pediatrics [Internet]. 2022 [cited 2023 Aug 9];10. Available from: https://www.frontiersin.org/articles/10.3389/fped.2022.908655
- Matsumoto J, Han B, Restrepo De Rovetto C, Welch T. Hypercalciuric Bartter syndrome: the resolution of nephrocalcinosis with indomethacin. American Journal of Roentgenology [Internet]. 1989 Jun 1 [cited 2023 Aug 9];152(6):1251–3. Available from: https://www.ajronline.org/doi/pdfplus/10.2214/ajr.152.6.1251
- Nuñez-Gonzalez L, Carrera N, Garcia-Gonzalez MA. Molecular basis, diagnostic challenges and therapeutic approaches of Bartter and Gitelman syndromes: a primer for clinicians. International Journal of Molecular Sciences [Internet]. 2021 Jan [cited 2023 Aug 10];22(21):11414. Available from: https://www.mdpi.com/1422-0067/22/21/11414
- Cunha T da S, Heilberg IP. Bartter syndrome: causes, diagnosis, and treatment. Int J Nephrol Renovasc Dis [Internet]. 2018 Nov 9 [cited 2023 Aug 10];11:291–301. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6233707/
- Johnson JR, Miller RS, Samuels P. Bartter syndrome in pregnancy. Obstetrics & Gynecology [Internet]. 2000 Jun [cited 2023 Aug 10];95(6 Part 2):1035. Available from: https://journals.lww.com/greenjournal/Citation/2000/06001/BARTTER_SYNDROME_IN_PREGNANCY.20.aspx#:~:text=With%20Bartter%20syndrome%2C%20there%20is,fetal%20growth%20restriction%2C%20and%20oligohydramnios.
- Francini F, Gobbi L, Ravarotto V, Toniazzo S, Nalesso F, Spinella P, et al. The dietary approach to the treatment of the rare genetic tubulopathies Gitelman’s and Bartter’s syndromes. Nutrients [Internet]. 2021 Aug 26 [cited 2023 Aug 10];13(9):2960. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8467039/




