


Introduction
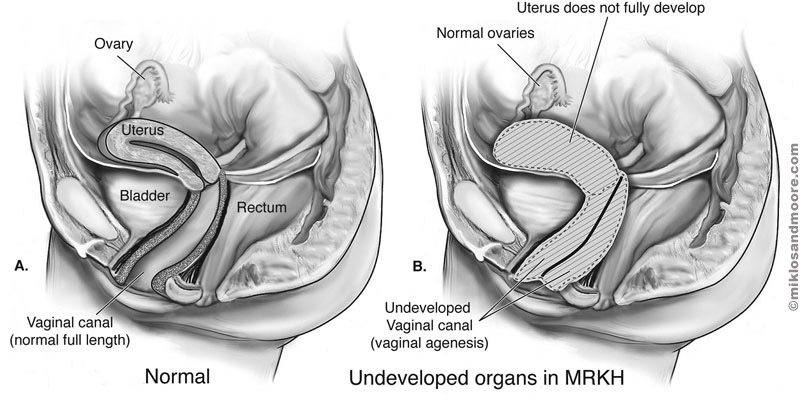
Vaginal agenesis is a rare condition where the vagina and the womb (uterus) are partially developed or are not developed at all.
There are several types of vaginal agenesis, such as the complete absence of the womb or the vagina, commonly known as Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome.
Vaginal agenesis is known to affect only 1 in 5000 people assigned female at birth (AFAB). This condition can be present since birth and unknown until symptoms occur later in life. Moreover, it can also be associated with skeletal or kidney problems.1
Causes and risk factors
The exact cause of the genetic condition is still not known, however, many congenital (present from birth) factors can lead to vaginal agenesis.
Genetic factors
People AFAB with vaginal agenesis will have the karyotype of 46 ‘XX’ chromosomes, meaning they will be female genetically. But there is very little evidence pointing to its inheritability.
Developmental abnormalities
In around 90% of vaginal agenesis cases, the individual will have MRKH syndrome.
Typically, the lower part of the Müllerian ducts develops into the womb and the vagina, while the upper part will develop into the fallopian tubes.
However, due to MRKH and the underdevelopment of the Müllerian ducts, both the vagina and the womb are either partially or completely absent.2
Types of vaginal agenesis
Isolated vaginal agenesis
This is where a urogenital tract malformation shows an absence of a vagina or having a vaginal ‘dimple’ that is shorter than 5 cm. The individual’s ovaries and their fallopian tubes will still function normally.3
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome
A disorder affecting the reproductive system that causes the vagina and the uterus to be either underdeveloped or absent, whilst the external genitalia appears normal.
When only the reproductive organs are affected it is classified as MRKH syndrome type 1. When other parts of the body are affected, such as the kidneys or skeletal system, then it is classified as MRKH syndrome type 2.4
Androgen insensitivity syndrome (AIS)
An individual who is assigned male at birth (AMAB) would typically have one X and one Y chromosome. This condition occurs when the X chromosome is resistant to androgens.
Therefore, the individual will have feminine physical traits but the genetic makeup of a person AMAB. AID can be characterised in 3 ways:
- Complete androgen insensitivity - the penis does not develop
- Partial androgen insensitivity - may have traits from either sex or both
- Mild androgen insensitivity - the penis develops but the body only partly responds to androgen5
Clinical presentation
Symptoms and signs
- Small pouch or dimple in the vaginal opening
- Lower abdominal pain (in the presence of a uterus with a missing connection to the vaginal canal)
- Atypical development of kidneys
- Skeletal abnormalities
- Hearing problems
- Amenorrhea
Typically, around 30 in 100 people with vaginal agenesis will have kidney abnormalities, commonly having a missing kidney or the dislocation of both organs. Though rare, both kidneys can also be joined, producing a ‘horseshoe-like’ shape. Around 12 in 100 people with vaginal agenesis will also show atypical skeletons, where two-thirds of them will have spine or rib defects.
It is important to understand the condition as it can affect the quality of life of the individual. This can include making it difficult to have sexual intercourse or having a baby. This condition can be recognised at birth, however, in most cases, it is not diagnosed until puberty.
Diagnostic evaluation
Vaginal agenesis is most commonly diagnosed during puberty when the menstrual periods have not started after the development of the breasts and body hair. Sometimes, a diagnosis can be made at an early age during an evaluation or a physical examination for other problems.
- Blood tests: They can be used to measure the hormone levels in the person’s blood for confirmation and to rule out any other conditions
- Imaging studies:
- MRI - This can give the paediatrician or gynaecologist a better, detailed image of the reproductive tract, and assess other organs around it
- Ultrasound - can show the health professional if the womb and the ovaries are present
- Genetic testing: Sometimes, karyotyping is performed for MRKH syndrome. This technique allows the professionals to assess the chromosomes in a sample of cells and pinpoint particular genetic causes for a condition. All individuals with MRKH have karyotype 46 XX
Management and treatment
If someone is diagnosed with vaginal agenesis, then the doctor will most likely not intervene immediately unless there is an urgent necessity, such as pain.
Non-surgical approaches
Vaginal dilators
This is considered the most efficient treatment for MRKH. It does not require surgery, however, it does require you to use the dilator once or twice a day or until the vaginal canal stretches to an adequate length.
The vaginal dilator is a hard, smooth plastic shaped like a tampon and it is used by applying pressure by hand. Over time, it stretches the vaginal opening.
Surgical procedures
Vaginoplasty
This is a technique that is utilised if vaginal dilation does not work for the individual. This technique uses surgery to form a functional vagina. Skin grafts are used in surgery to form a functional vagina. Skin and tissue can be taken from other parts of the body such as the outer thigh and buttocks.
Several types of vaginoplasty can be performed:
- McIndoe vaginoplasty - The surgeon would make an incision to create an opening and place the tissue graft over a mould to form a vagina, connecting it to a formed canal
- Intestinal vaginoplasty - This is where part of the colon’s tissue is used to make a vaginal opening and a new vagina
- Peritoneal vaginoplasty - This type uses the lining of the abdominal cavity to create the vagina
Neovaginal construction
This is also known as the Vecchietti procedure and it involves using an olive-shaped bead in the vaginal opening, which is connected to a separate traction in the lower abdomen or connected through the naval.6
Complications and risks
Surgical risks
Post-surgery care is incredibly important to the individual’s recovery. To maintain results, the patient typically must use the vaginal dilator for 3-6 months, because it can help to stretch the vaginal opening. This will vary depending on the type of procedure performed.
One of the main risks involved with surgery is vaginal stenosis, which occurs when the vagina becomes narrower or shorter.
Psychological and emotional challenges
Being diagnosed and coping with vaginal agenesis can be very difficult for anyone, but especially for adolescents. It is important for the patient to have a psychologist or social worker working alongside the treatment team to help cope with the challenging aspects of the condition, such as the possibility of infertility.
It can also be helpful to reach out and connect with support groups aimed at supporting other individuals with the same condition.
Long-term considerations
Vaginal agenesis can have an impact on the sexual relationship of an individual, causing them to have a lot of pain and discomfort during sex. After treatment, the person can have sexual activity.
People with missing or partially developed wombs cannot get pregnant. If the ovaries are healthy, however, it is possible to have a baby through different fertility methods, for instance, IVF or gestational surrogacy.
Future directions in research and treatment
Due to several complications with the current techniques and surgical methods, researchers are always looking for ways to optimise the treatment to better suit the individual with the condition.
Advances in surgical techniques
Currently, the use of neovaginal procedures has a few complications, such as the snapping of the mesh or rupture of tissue, among others.
As a result, research has developed a new laparoscopic Vecchietti-based procedure, considered to be low-risk as it requires minimal care to maintain the long-term effects.
Similarly, a recent study suggests that patients with MRKH syndrome undergo uterine transplantation, as studies showed mutations and epigenetic changes, hoping to help patients deliver a biological child.7,8
Fertility preservation options
There are several options for fertility preservation available. If the ovaries are normal, it is possible to have the eggs frozen.
Psychosocial support innovations
With vaginal agenesis being a difficult condition to live with, many psychological support groups can help the patient to better understand the condition and connect to others that also have it.
Summary
Vaginal agenesis, being a rare condition which affects a small number of females worldwide, has a lot of complicated effects of which the cause is still unknown. It is known to affect the female reproductive system, though it can be partially developed or not developed at all, causing complications such as infertility and lower abdominal pain. Currently, vaginal dilators and surgery are the best options for patients, however, research on treatment can be improved as there are various disadvantages to the surgeries.
References
- ACOG Committee on Adolescent Health Care. ACOG Committee Opinion No. 355: Vaginal agenesis: diagnosis, management, and routine care. Obstet Gynecol. 2006 Dec;108(6):1605–9.
- Ray U, Adhikari S, Dhital R, Shrestha S, Shah S, Poudel S, et al. Mayer-rokitansky-kuster-hauser syndrome: a rare case report from nepal. Annals of Medicine and Surgery [Internet]. 2022 Oct 1 [cited 2023 Sep 20];82:104725. Available from: https://www.sciencedirect.com/science/article/pii/S2049080122014856
- Slaoui A, Benzina I, Talib S, Etber A, Zeraidi N, Lakhdar A, et al. Congenital vaginal atresia: about an uncommon case. Pan Afr Med J [Internet]. 2020 Sep 17 [cited 2023 Sep 20];37:69. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7680234/
- Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (Mrkh) syndrome: a comprehensive update. Orphanet J Rare Dis [Internet]. 2020 Aug 20 [cited 2023 Sep 20];15:214. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7439721/
- Gottlieb B, Trifiro MA. Androgen insensitivity syndrome. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cited 2023 Sep 20]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1429/
- Miller PB, Forstein DA. Creation of a neovagina by the vecchietti procedure in a patient with corrected high imperforate anus. JSLS [Internet]. 2009 [cited 2023 Sep 20];13(2):221–3. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3015920/
- Mikos T, Gordts S, Grimbizis GF. Current knowledge about the management of congenital cervical malformations: a literature review. Fertility and Sterility [Internet]. 2020 Apr 1 [cited 2023 Sep 20];113(4):723–32. Available from: https://www.sciencedirect.com/science/article/pii/S0015028220300984
- Brucker SY, Gegusch M, Zubke W, Rall K, Gauwerky JF, Wallwiener D. Neovagina creation in vaginal agenesis: development of a new laparoscopic Vecchietti-based procedure and optimized instruments in a prospective comparative interventional study in 101 patients. Fertility and Sterility [Internet]. 2008 Nov 1 [cited 2023 Sep 20];90(5):1940–52. Available from: https://www.sciencedirect.com/science/article/pii/S0015028207034668




